
Tri de lignage incomplet et taille d’une population ancestrale
Dans le dernier billet de cette série, nous avons introduit le (difficile) concept de l’arbre généalogique génétique discordant avec un arbre généalogique des espèces à cause d’un tri de lignées incomplet (TLI). Dans ce billet, nous examinerons l’une des implications intéressantes du TLI – son utilisation pour estimer la taille d’une population – avant de continuer sur d’autres sujets. (Encore une fois, si ce sujet semble trop difficile, n’hésitez pas à le mettre de côté avec le billet précédent ; les billets suivants de cette série ne dépendront pas de la compréhension de cette question.)
Nous pouvons utiliser le TLI pour mesurer la taille d’une population parce que les arbres discordants nous donnent une façon de mesurer le nombre d’allèles présents dans une population ancestrale (ce qui peut ensuite être utilisé pour estimer le nombre d’individus dans cette population). Avant d’en arriver aux détails, cependant, revoyons brièvement dans quelle mesure la spéciation est un phénomène qui se passe à l’échelle de la population.
Nous avons vu dans les billets précédents de cette série que les événements de spéciation commencent lorsque deux populations deviennent génétiquement isolées l’une de l’autre (soit complètement, soit partiellement). Cela a pour conséquence une divergence de la moyenne des caractéristiques des deux populations, ce qui peut conduire par la suite et à long terme à une spéciation. Ce qu’il nous faut souligner ici, c’est que les deux populations sont des populations – un groupe d’organismes d’une même espèce qui se reproduisent entre eux. Comme nous l’avons vu, les populations sont capables de transmettre une diversité génétique bien plus importante qu’un individu : là où un individu ne peut porter que deux allèles d’un gène donné, une population peut en maintenir des centaines voire des milliers.
Arbres discordants – une fenêtre ouverte sur le passé.
En gardant cela à l’esprit, nous pouvons revenir à notre réflexion sur le tri de lignées incomplet et sur les arbres généalogiques génétiques discordants avec un arbre généalogique des espèces qui en résultent. L’exemple que nous avons utilisé montrait des gorilles et des chimpanzés possédant des allèles plus proches, tandis que l’allèle humain était plus éloigné :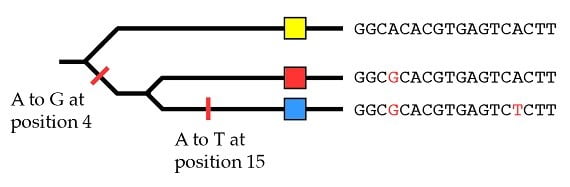
Nous avons ensuite décrit un exemple de tri de lignage incomplet où les gorilles et les chimpanzés héritent des allèles les plus proches et les humains d’allèles plus éloignés :
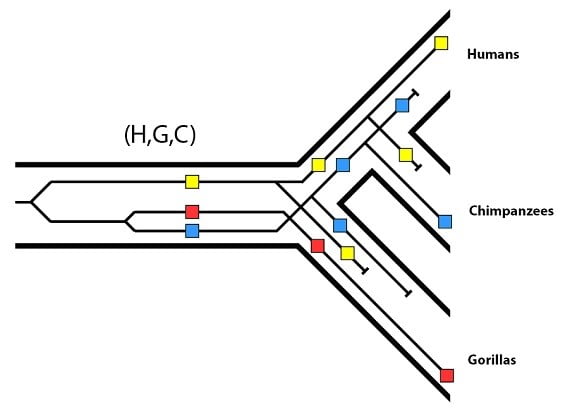
Nous sommes désormais prêts à voir ce que nous pouvons inférer de ce patron, et ce qu’il nous dit de la population ancestrale commune (H,G,C). D’abord, ce patron nous dit que les allèles bleu et rouge étaient présents avant la séparation des lignages du chimpanzé et du gorille. Puisque nous savons grâce à l’arbre généalogique des espèces que la population ancestrale commune (gorille/chimpanzé) correspond à la population ancestrale commune (H,G,C), cela confirme que les allèles bleu et rouge faisaient partie de la variation que cette population a maintenu. La deuxième chose à remarquer, c’est que l’allèle jaune est plus ancestral ; en d’autres termes, il a moins de mutations que les allèles rouge et bleu. Cela signifie que l’allèle jaune est plus vieux que les allèles rouge ou bleu. Cela place l’allèle jaune avant l’événement de spéciation (G) / (H,C) sur la phylogénie. De plus, puisque les humains ont l’allèle jaune, il doit avoir été présent dans la population ancestrale commune (H,C) au moment où elle s’est séparée du lignage (G). Si nous rassemblons ces deux informations, cela signifie que l’allèle jaune était aussi présent dans la population (H,G,C). En l’absence de nouvelles mutations (qui sont exclues dans ces analyses) ce patron ne peut être produit que par la présence des trois allèles dans la population (H,G,C). Bien que les espèces d’aujourd’hui ne possèdent chacune qu’un allèle, on peut inférer que leur population ancestrale partagée avait les trois.
Les arbres généalogiques génétiques discordants sont donc une fenêtre ouverte sur le passé qui révèle la diversité génétique d’une population ancestrale, c’est-à-dire le nombre d’allèles qu’elle maintenait pour une région donnée du génome. En comparant de larges sections des données du génome des humains, des chimpanzés et des gorilles, il est possible d’avoir une estimation exacte de la taille de la population ancestrale (H,G,C) (environ 50000 individus). Cette mesure, que l’on appelle la taille effective de la population (notée Ne), est la taille de la population dont on a besoin pour transmettre la quantité observée de variation génétique d’une population ancestrale à aujourd’hui. Le lignage commun ancestral humain/chimpanzé (H,C), estimé par l’usage des mêmes méthodes, aurait aussi eu environ 50000 individus dans son histoire.
Test du modèle avec une espèce en plus : le génome de l’orang-outan.
La séquence du génome de l’orang-outan (terminée en 2011) a donné aux chercheurs l’opportunité de vérifier ces estimations en utilisant un ensemble de données en plus. Le lignage de l’orang-outan se détache de la phylogénie primate d’une population ancestrale commune (la population (H,O,G,C), « O » désignant l’orang-outan), reste donc la population ancestrale (H,G,C), qui passera par la spéciation plus tard :
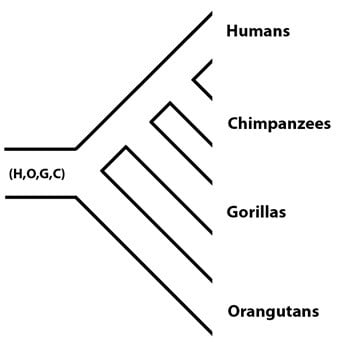
En utilisant des estimations faites pour les tailles des populations (H,G,C) et (H,C), les chercheurs ont pu prédire qu’une petite fraction des génomes de l’humain et de l’orang-outan seraient plus proches, c’est-à-dire que le tri de lignées incomplet devrait avoir produit de rares régions du génome dans lesquelles les allèles de l’humain et de l’orang-outan sont plus similaires qu’avec ceux d’autres primates. La valeur attendue de telles régions correspondantes de (H,O) est petite (environ 1.2%) lorsqu’on la compare à la valeur prédite pour les régions correspondantes de (H,G) (environ 25%), en grande partie parce que les humains, les chimpanzés et les gorilles sont passés par la spéciation dans une période temporelle relativement courte, alors que le temps passé entre la divergence de l’orang-outan et celle du gorille est plus long. La fraction de notre génome qui correspond de plus près au génome de l’orang-outan est d’environ 0.8%, un résultat remarquablement proche de la valeur prédite, et cohérent avec les valeurs Ne estimées pour les populations (H,G,C) et (H,C) dans des travaux antécédents. En d’autres termes, lorsqu’on compare les génomes de primates, nous voyons un patron de tri de lignées incomplet, conforme à l’attendu : notre génome correspond à celui des chimpanzés le plus fréquemment, puis à celui du gorille, puis à celui de l’orang-outan. (Soit dit en passant, il est formellement possible qu’une fois que le génome du gibbon aura été séquencé et analysé, il y ait une trace de tri de lignées incomplet pour donner des regroupements des allèles (humain, gibbon), mais il est probable que cette fraction du génome soit trop petite pour être détectée avec fiabilité, puisque les gibbons se détachent de l’arbre des primates bien avant les orang-outans).
En résumé, et pour la suite.
Loin d’être un « problème » pour l’ancêtre commun, le tri de lignées incomplet est une conséquence attendue de populations qui passent par des événements de spéciation, et une fenêtre ouverte sur leur diversité génétique passée. Le résultat final dans une phylogénie, comme nous l’avons vu, est un sous-ensemble de caractéristiques qui ont un arbre généalogique discordant avec l’arbre généalogique des espèces. Dans le prochain billet de cette série, nous explorerons un autre phénomène que révèlent les patrons contradictoires avec un arbre généalogique des espèces: l’évolution convergente.
Pour en savoir plus, lectures complémentaires.
Hobolth A, et al., (2007). Genomic Relationships and Speciation Times of Human, Chimpanzee, and Gorilla Inferred from a Coalescent Hidden Markov Model. PLoS Genet 3(2): e7 (source)
Holboth A., et al. (2011). Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Research. 2011 March; 21(3) 349. (source)
48 Articles pour la série :
- 01-L'évolution expliquée : Introduction
- 02-L'Evolution : Une théorie testée, pas une simple hypothèse !
- 03-Biogéographie
- 04-Une introduction à la variation, à la sélection naturelle et artificielle
- 05-Les chiens domestiques
- 06-Comment ça marche, la sélection naturelle ?
- 07-La sélection naturelle et le lignage humain.
- 08-Les bases de la variation héréditaire, première partie
- 09-Les bases de la variation héréditaire, deuxième partie.
- 10-De la variation à la spéciation (1)
- 11-De la variation à la spéciation (2)
- 12-De la variation à la spéciation 3
- 13-De la variation à la spéciation (4)
- 14-Les génomes sont comme des anciens textes (1)
- 15-Les génomes comparés aux textes anciens (2)
- 16-Les génomes comparés aux textes anciens (3): les origines de l'homme
- 17-Le génome comparé à un texte ancien (4)
- 18-Le génome comparé à un texte ancien (5): rattacher le tout ensemble.
- 19-Les arbres généalogiques des espèces, des gènes, et tri incomplet des lignées
- 20-Tri de lignage incomplet et taille d’une population ancestrale (Cet article)
- 21-Une introduction à l’homoplasie et à la convergence évolutive
- 22-Evolution convergente et homologie profonde.
- 23-Coévolution et la course à l’armement prédateur / proie
- 24-Le parasitisme, le mutualisme et la co-spéciation
- 25-Comprendre l’endosymbiose
- 26-La diversification cambrienne et la mise en place des plans d’organisation animaux. Première partie.
- 27-La diversification cambrienne et la mise en place des plans d’organisation animaux. 2e partie.
- 28-La mise en place des plans d’organisation des corps vertébrés, première partie.
- 29-La mise en place des plans d’organisation des corps vertébrés, deuxième partie.
- 30-La mise en place des plans d’organisation des vertébrés : Troisième partie.
- 31-La mise en place des plans d’organisation des vertébrés, quatrième partie.
- 32-Des reptiles aux mammifères.
- La révolution placentaire : deuxième partie.
- Du primate à l’humain, première partie.
- Du primate à l’humain, deuxième partie
- Du primate à l'humain (3)
- La paléontologie hominienne : une petite esquisse des preuves actuelles
- Devenir humain (1) : Eve mitochondriale et Adam Y-chromosomique
- Analogie entre évolution biologique et évolution du langage
- Devenir humain (3) : paléogénomiques et la toile emmêlée de la spéciation humaine
- 41-L’évolution, Partie 1: frontières scientifiques, abiogenèse et apologétique chrétienne
- 42-Aux frontières de l’évolution, Partie 2: l’abiogenèse et la question du naturalisme
- 43-Aux frontières de l’évolution, Partie 3 : l’hypothèse du monde à ARN
- 44-Aux frontières de l’évolution, partie 4 : Contingence versus convergence
- 45-Aux frontières de l’évolution, partie 5 : Contigence versus convergence dans l’expérience LTEE
- 46- L’évolution et le chrétien, première partie: Est-ce que l’évolution est un mécanisme sans but ?
- 47- L’évolution et le chrétien, deuxième partie: Une créature merveilleuse
- 48- L’évolution et le chrétien, partie 3 : Dire la vérité dans l’amour


