
Dans les derniers billets de cette série, nous avons examiné le patron global que l’on voit en comparant des génomes proches les uns des autres, et nous avons vu que les données multiples forment le même arbre généalogique ou phylogénie. Dans ce billet, nous allons chercher à comprendre plus profondément les phylogénies et plus particulièrement dans quelle mesure nous nous attendons à ce que certaines caractéristiques des génomes entrent en contradiction avec leurs arbres généalogiques.
Mais d’abord, une brève parenthèse : ce sujet est compliqué, il sera peut-être difficile à comprendre au début. Pourtant, si vous êtes arrivés jusqu’ici dans cette série, vous avez déjà les outils nécessaires pour comprendre et avec un effort, vous pourrez comprendre plus profondément qu’avant les génomes parents. Si ce sujet particulier reste tout de même obscur, ne vous inquiétez pas – le reste de cette série ne dépendra pas de la compréhension de ce point plus subtil. N’hésitez pas à poser des questions dans les commentaires si certaines choses ne sont pas claires.
Arbres généalogiques des espèces.
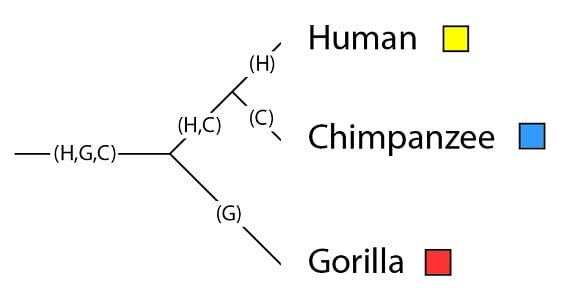
Revenons à un exemple qui nous est maintenant familier, celui des humains, des chimpanzés et des gorilles :
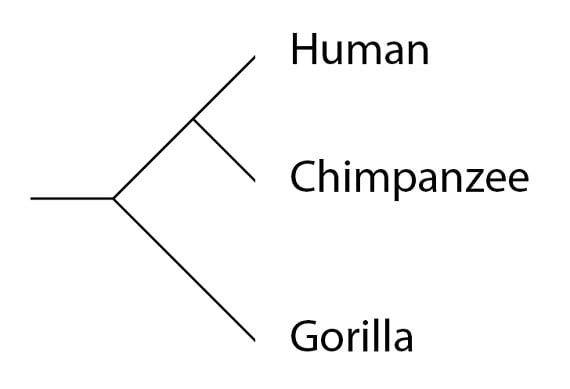
On appelle aussi les phylogénies des “arbres généalogiques des espèces” puisque “arbre généalogique” est un autre nom pour phylogénie. Un arbre généalogique des espèces nous montre un patron global des espèces qui ont partagé un ancêtre commun il y a plus ou moins longtemps. En d’autres termes, comme nous l’avons dit dans le dernier billet de cette série, une phylogénie est une mesure de l’histoire partagée et distincte de deux espèces. Plus les deux espèces partagent une histoire commune, plus on s’attend à ce que, en moyenne, elles soient similaires. Les humains et les chimpanzés, par exemple, continuent à partager une histoire pendant plusieurs millions d’années après s’être séparés de la population commune aux gorilles. Cette histoire commune est ce qui, en moyenne, rend les génomes de l’humain et du chimpanzé plus similaires l’un à l’autre qu’au génome du gorille. Les gènes individuels (et leurs allèles) peuvent avoir une histoire différente à l’intérieur des espèces alors qu’elles se séparent. Pour ce type d’analyse, nous devons examiner des phylogénies de gènes individuels, ce qu’on appelle des « arbres généalogiques génétiques ».
Arbres généalogiques génétiques
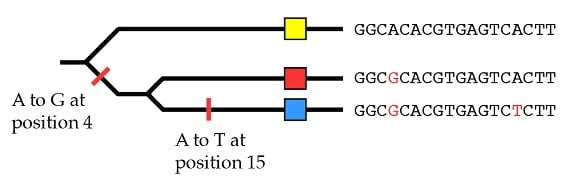
Si vous vous rappelez les billets précédents qui expliquaient comment la variation (d’allèles) survenait par la mutation, on peut comprendre intuitivement que les principes qui sont utilisés pour regrouper des espèces en une phylogénie puissent aussi être utilisés pour regrouper des allèles d’un gène en une phylogénie. Par exemple, regardez la séquence ADN de trois allèles du même gène, que nous pouvons représenter par les allèles jaune, rouge et bleu (les boîtes colorées). Les différences de séquences qui distinguent ces allèles sont les lettres surlignées en rouge :
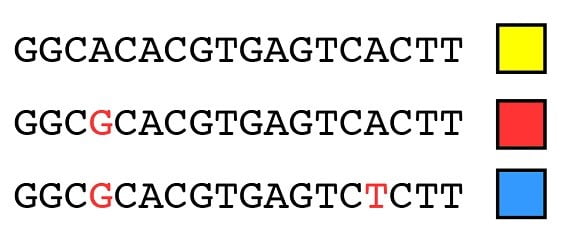
En utilisant les mêmes principes que pour les espèces, nous pouvons expliquer l’origine de ces trois allèles par deux événements de mutation (en postulant que l’allèle jaune est l’état ancestral) :
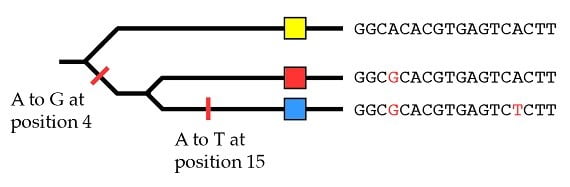
Donc à l’intérieur d’une population, nous pouvons reconstruire l’histoire de l’allèle d’un gène individuel en utilisant les mêmes méthodes que celles appliquées précédemment à des espèces entières.
La spéciation accompagnée d’une variation génétique (ou pas)
Donc la mutation produit constamment de nouveaux allèles (de nouvelles variations) à l’intérieur des populations, et des processus tels que la sélection naturelle et la dérive génétique contribuent soit à augmenter soit à diminuer la fréquence des allèles dans les populations au cours du temps. Nous avons passé du temps à expliquer comment les événements de spéciation, à commencer par les populations qui se séparent et se différencient de plus en plus au cours du temps, conduisent à la formation d’espèces distinctes. Il nous reste à rassembler ces idées : à considérer ce qui pourrait arriver à la variation (aux allèles) dans une population alors qu’elle traverse un événement de spéciation.Pour cela, cherchons nos allèles hypothétiques à travers les événements de spéciation qui ont conduit aux humains, aux chimpanzés et aux gorilles.
Cet arbre généalogique des espèces a les populations suivantes : la population ancestrale commune aux trois espèces, désignée par (H,G,C) pour (Humain, Gorille, Chimpanzé) ; la population ancestrale commune aux humains et aux chimpanzés (H,C), et les lignages qui ont conduit aux espèces d’aujourd’hui après le dernier événement de spéciation sur la phylogénie (H), (G), (C) :
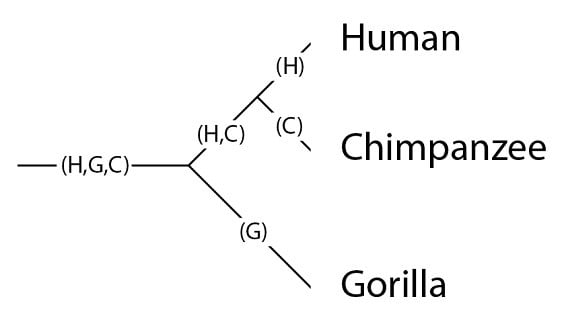
Il est important de garder à l’esprit qu’une seule ligne sur la phylogénie correspond à une population, et que les populations peuvent avoir des variations génétiques. Plaçons nos trois allèles dans la population (H,G,C) :
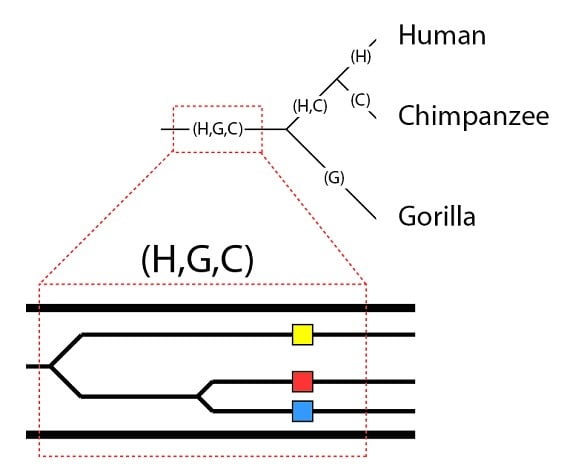
Nous sommes désormais prêts à explorer comment ces allèles seront hérités (ou pas) à travers les événements de spéciation. Une première possibilité est directe : chacun des trois allèles est hérité par une espèce différente. Cette possibilité s’appelle « tri complet des lignages ».. Si cela est possible, ce n’est pas du tout certain surtout si des plusieurs évènements de spéciation se produisent sur une courte période de temps. On peut ainsi avoir maintien des différents allèles, maintien d’un polymorphisme dit ancestral, dans les différents lignages. Il peut y avoir perte d’allèles dans chaque lignée via la sélection naturelle ou la dérive génétique qui peut conduire à une phylogénie « discordante » avec celle des espèces. C’est ce qu’on appelle le « le tri incomplet des lignages», et pour un grand génome, il est évident qu’au moins certains gènes manifesteront ce phénomène.
Distribution incomplète dans le lignage
La première difficulté que rencontreront les trois allèles pour se distribuer complètement dans les lignages sera l’événement de spéciation qui sépare les lignages (H,C) et (G). Vous vous rappellez que c’est un exemple de l’effet fondateur – un échantillon sélectionné par hasard qui peut exclure des allèles d’une nouvelle sous-population par chance :
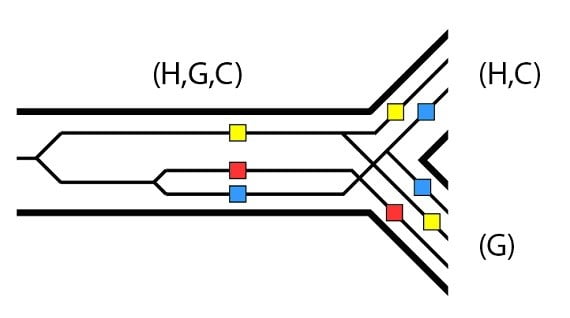
Examinons un scénario possible à partir de l’événement de spéciation entre (H,C) et (G). Dans le lignage (G), les allèles jaune et bleu sont perdus au cours du temps. Lors de l’événement de spéciation qui sépare (H) et (C), les allèles jaune et bleu vont dans les deux lignages, mais dans le lignage (C), l’allèle jaune est perdu plus tard. De même, l’allèle bleu est perdu plus tard dans le lignage (H) :
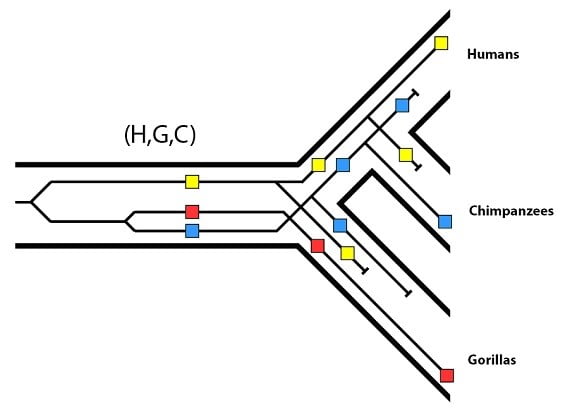
Pour ce gène particulier, nous avons donc le patron suivant :
Enfin, nous voyons le problème : l’arbre généalogique génétique pour ces allèles entre en contradiction avec l’arbre généalogique des espèces. Rappelez-vous que dans l’arbre généalogique génétique, les allèles rouge et bleu sont plus proches qu’avec l’allèle jaune :
Dans l’arbre généalogique des espèces, cependant, les deux espèces parentes les plus proches (les chimpanzés et les humains) n’ont pas les allèles les plus proches, elles ont des allèles qui présentent le plus de différences.
Maintenant que nous avons travaillé cet exemple, la raison derrière ce décalage (espérons-le) est claire : il n’y a aucune garantie que les allèles se distribuent dans un lignage pour correspondre au patron global des espèces. Si un gène varie dans une population qui traverse un événement de spéciation, on s’attend à ce que parfois il soit assorti avec un patron qui ne correspond pas à celui des espèces ; dans certains cas, il aura un arbre généalogique génétique qui sera « discordant » avec l’arbre généalogique des espèces. Pour une population qui a des milliers de gènes avec de multiples allèles, il est évident que certains allèles formeront un patron discordant. Loin d’être un problème pour la théorie de l’évolution, les arbres généalogiques discordants sont prédits par elle. Ce serait un problème si nous ne les observions pas ; mais de fait, nous les observons, et comme nous le verrons la prochaine fois, nous les observons précisément dans le patron qui correspond à ce à quoi l’on s’attend en se basant sur les arbres généalogiques des espèces.
Dans le prochain billet de cette série, nous verrons comment les arbres généalogiques génétiques discordants peuvent être utilisés pour déterminer une autre caractéristique qui intéresse les scientifiques : les tailles de population des lignages sur une phylogénie.
48 Articles pour la série :
- 01-L'évolution expliquée : Introduction
- 02-L'Evolution : Une théorie testée, pas une simple hypothèse !
- 03-Biogéographie
- 04-Une introduction à la variation, à la sélection naturelle et artificielle
- 05-Les chiens domestiques
- 06-Comment ça marche, la sélection naturelle ?
- 07-La sélection naturelle et le lignage humain.
- 08-Les bases de la variation héréditaire, première partie
- 09-Les bases de la variation héréditaire, deuxième partie.
- 10-De la variation à la spéciation (1)
- 11-De la variation à la spéciation (2)
- 12-De la variation à la spéciation 3
- 13-De la variation à la spéciation (4)
- 14-Les génomes sont comme des anciens textes (1)
- 15-Les génomes comparés aux textes anciens (2)
- 16-Les génomes comparés aux textes anciens (3): les origines de l'homme
- 17-Le génome comparé à un texte ancien (4)
- 18-Le génome comparé à un texte ancien (5): rattacher le tout ensemble.
- 19-Les arbres généalogiques des espèces, des gènes, et tri incomplet des lignées (Cet article)
- 20-Tri de lignage incomplet et taille d’une population ancestrale
- 21-Une introduction à l’homoplasie et à la convergence évolutive
- 22-Evolution convergente et homologie profonde.
- 23-Coévolution et la course à l’armement prédateur / proie
- 24-Le parasitisme, le mutualisme et la co-spéciation
- 25-Comprendre l’endosymbiose
- 26-La diversification cambrienne et la mise en place des plans d’organisation animaux. Première partie.
- 27-La diversification cambrienne et la mise en place des plans d’organisation animaux. 2e partie.
- 28-La mise en place des plans d’organisation des corps vertébrés, première partie.
- 29-La mise en place des plans d’organisation des corps vertébrés, deuxième partie.
- 30-La mise en place des plans d’organisation des vertébrés : Troisième partie.
- 31-La mise en place des plans d’organisation des vertébrés, quatrième partie.
- 32-Des reptiles aux mammifères.
- La révolution placentaire : deuxième partie.
- Du primate à l’humain, première partie.
- Du primate à l’humain, deuxième partie
- Du primate à l'humain (3)
- La paléontologie hominienne : une petite esquisse des preuves actuelles
- Devenir humain (1) : Eve mitochondriale et Adam Y-chromosomique
- Analogie entre évolution biologique et évolution du langage
- Devenir humain (3) : paléogénomiques et la toile emmêlée de la spéciation humaine
- 41-L’évolution, Partie 1: frontières scientifiques, abiogenèse et apologétique chrétienne
- 42-Aux frontières de l’évolution, Partie 2: l’abiogenèse et la question du naturalisme
- 43-Aux frontières de l’évolution, Partie 3 : l’hypothèse du monde à ARN
- 44-Aux frontières de l’évolution, partie 4 : Contingence versus convergence
- 45-Aux frontières de l’évolution, partie 5 : Contigence versus convergence dans l’expérience LTEE
- 46- L’évolution et le chrétien, première partie: Est-ce que l’évolution est un mécanisme sans but ?
- 47- L’évolution et le chrétien, deuxième partie: Une créature merveilleuse
- 48- L’évolution et le chrétien, partie 3 : Dire la vérité dans l’amour



