
Les génomes comparés à des textes copiés : rattacher le tout ensemble.
Lors des derniers billets de cette série, nous avons examiné les patrons généraux que l’on observe lorsqu’on compare les génomes d’espèces distinctes. Nous avons vu que le patron que nous observons est parfaitement cohérent dans les espèces qui partagent un ancêtre commun – et que leurs génomes sont des copies modifiées de ce qui a été autrefois le génome de leur population ancestrale. Ce que nous n’avons pas encore vu, en revanche, c’est que les lignes d’évidence que nous avons examinées – comparaisons des structures des génomes, séquences de gènes fonctionnels, mutations spécifiques conduisant à des gènes désactivés – forment un patron cohérent. Une bonne théorie se construit sur la cohésion d’une multiplicité de lignes d’évidence indépendantes, et la génomique comparative soutient très fortement la théorie de l’évolution.
D’abord, les “fautes de frappe” partagées.
Revenons à l’exemple des récepteurs olfactifs désactivés que nous avons vu dans notre dernier billet. En nous fondant sur ce petit ensemble de gènes défectueux, nous avons construit « l’arbre généalogique » des humains, chimpanzés, gorilles, et orang-outans :
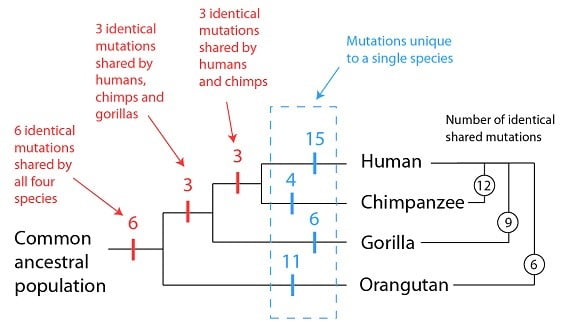
Proposer un arbre généalogique d’espèces parentes – ou pour introduire un terme technique, une phylogénie – est une façon graphique à la fois (a) de représenter un grand ensemble de données et (b) de proposer une hypothèse sur l’histoire de ces données. Dans cet ensemble de données, nous avons deux catégories de caractéristiques à expliquer : les mutations identiques chez plus d’une espèce, et les mutations uniques à une seule espèce. Comme nous l’avons montré auparavant, le phylogénie ci-dessus correspond très bien aux données ; les patrons des événements partagés et des événements uniques sont représentés par la même phylogénie. Les événements partagés arrivent une fois, dans l’ancêtre commun, et les événements uniques arrivent une fois que les deux espèces se sont séparées.
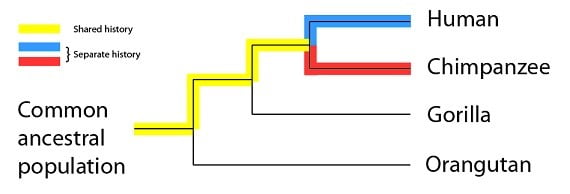
L’hypothèse que cette phylogénie propose, c’est que les histoires de ces quatre espèces sont partagées ou séparées en proportions différentes. Par exemple, les humains et les chimpanzés auraient l’histoire partagée la plus longue de ces quatre espèces (surlignée en jaune), et par comparaison des histoires séparées plus courtes (surlignées en bleu et rouge) :
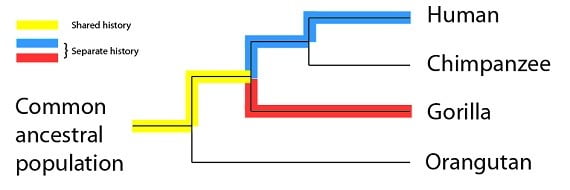
Les humains et les gorilles, cependant, auraient une histoire partagée plus courte (et une histoire séparée plus longue) sur la même période de temps :
Enfin, les orang-outans et les humains auraient une histoire partagée encore plus courte que celles des autres primates, et une histoire séparée plus longue :
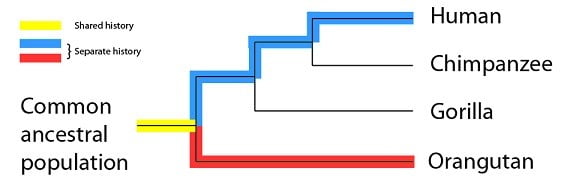
Donc à partir de cet ensemble relativement petit de données (une poignée de mutations partagées par quelques gènes) nous avons une hypothèse détaillée des espèces qui partagent le plus une histoire commune – une hypothèse que nous pouvons tester en utilisant d’autres lignes d’évidence.
Des fautes de frappe aux phrases.
Maintenant que nous avons utilisé un petit ensemble des « fautes de frappe partagées » que l’on trouve dans ces quatre génomes pour assembler une phylogénie, nous pouvons considérer ce que cette phylogénie prédirait en comparant les séquences de gènes individuels chez ces quatre espèces. La clé, c’est la partie d’histoire que deux espèces partagent dans une phylogénie – alors que ce qui deviendra plus tard deux espèces n’est encore qu’une population, avec un génome commun. Elles auront la même séquence pour tout gène donné (aux variations à l’intérieur de la population près). Plus l’histoire que deux espèces partagent est longue, plus nous nous attendrons à voir les séquences de leurs gènes similaires. Plus leur histoire séparée est longue, plus nous nous attendrons à voir des gènes différents, à cause d’événements mutationnels qui arrivent dans la partie de la phylogénie de leur « histoire séparée ».
Grâce à la séquence du génome de l’orang-outan en 2011 et du gorille en 2012, nous pouvons désormais estimer cette prédiction en utilisant un très large ensemble de données pour les quatre espèces. Les séquences de l’homme et du chimpanzé sont quasiment identiques statistiquement (identiques à 98,6%) ; celles de l’humain et du gorille un peu moins (identiques à 98,3%) ; et celles des humains et des orang-outans encore moins (identiques à 96,6%). Ces résultats correspondent au patron prédit :

En d’autres termes, utiliser de longues séquences d’un génome pour assembler une phylogénie de ces espèces produit la même phylogénie que celle que produisent les données des mutations partagées. Le patron requis par les données de mutations partagées (un petit ensemble des séquences ADN de ces espèces) est aussi celui qui explique le mieux l’identité globale du génome que l’on observe.
Des phrases aux chapitres
Avec les données de mutations partagées et les données de la séquence globale soutenant la même phylogénie, nous pouvons aller plus loin et tester cette hypothèse en utilisant la structure du génome – l’organisation spatiale des gènes sur les chromosomes, ou « chapitres » pour reprendre notre analogie du livre copié. Pour ce qui est des données des mutations partagées, les similitudes et différences que nous observons devraient être soit des caractéristiques partagées (du patron prédit) soit des caractéristiques uniques (survenues dans une espèce une fois, et qui l’ont séparée des autres). Globalement, lorsqu’on compare la structure des chromosomes des quatre espèces, les résultats répondent à nos attentes. Lorsque nous comparons la structure des chromosomes, celle des humains est plus similaire à celle des chimpanzés, un peu moins à celle des gorilles, et encore moins à celle des orang-outans, comme attendu. Pour illustrer ce patron d’un exemple spécifique, revenons à la différence structurelle majeure du chromosome entre les humains et les grands singes, l’événement de fusion qui a conduit au chromosome humain numéro 2. Comme nous l’avons déjà montré, le chromosome fusionné est présent dans les humains mais pas chez les chimpanzés, ni les gorilles, ni les orang-outans, ce qui signifie que cet évènement s’est produit après la séparation du lignage humain de celui du chimpanzé. Un examen plus précis de cette région chez les gorilles et les orang-outans révèle une différence de plus : une partie de cette région est inversée dans les génomes de l’humain et du chimpanzé (encadrée en rouge) quand on les compare avec la région équivalente chez les gorilles et les orang-outans (encadrée en bleu) :
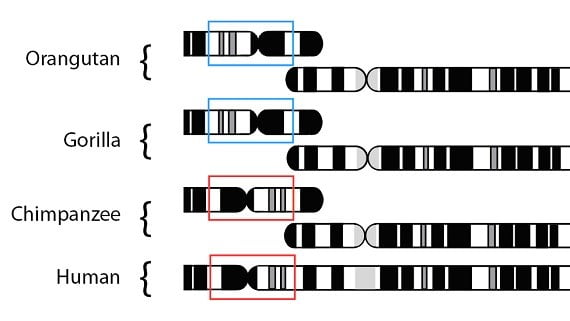
Les humains sont plus similaires aux chimpanzés (la plupart des régions correspondent), et le sont moins aux autres singes, comme on s’y attendait. Les différences que l’on voit sont aussi facilement représentées sur la phylogénie formée par les autres lignes d’évidence. Puisque l’événement d’inversion est commun aux humains et aux chimpanzés (mais il n’est pas présent chez les autres espèces), il a probablement eu lieu dans la population commune des humains et des chimpanzés après s’être séparée du lignage qui a conduit aux gorilles. L’événement de fusion aurait lieu plus tard, sur le lignage qui a conduit aux humains (et comme nous l’avons vu, il serait partagé aussi par d’autres espèces de parenté plus proche que les grands singes avec les humains.) Comme nous nous y attendons, la phylogénie prédite en utilisant seulement les données structurelles des chromosomes correspond à la phylogénie prédite par d’autres lignes d’évidence :
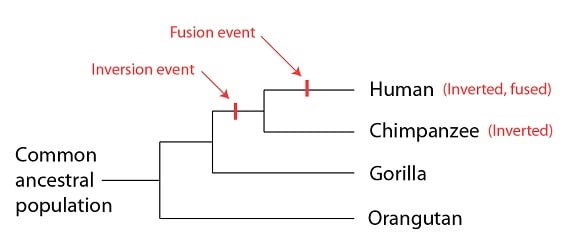
En résumé
Comme nous l’avons vu au début de cette série, une bonne théorie (au sens scientifique du terme) est une théorie qui est soutenue par une multiplicité de lignes d’évidence et qui fait des prédictions exactes. Grâce à l’avènement de la génomique comparative moderne, la théorie de l’évolution s’est révélée plus robuste que ce que Darwin aurait imaginé. Nous pouvons dire avec confiance que nous partageons des ancêtres avec d’autres espèces et que cette conclusion n’est pas prête de changer, alors même que l’on a de nouvelles informations.
Dans le prochain billet de cette série, nous tournerons notre attention vers les caractéristiques qui ne correspondent pas parfaitement aux phylogénies prédites. Loin d’être un problème (comme les opposants à l’évolution le prétendent souvent), ces caractéristiques sont une source riche d’information qui révèle plus encore notre passé.
48 Articles pour la série :
- 01-L'évolution expliquée : Introduction
- 02-L'Evolution : Une théorie testée, pas une simple hypothèse !
- 03-Biogéographie
- 04-Une introduction à la variation, à la sélection naturelle et artificielle
- 05-Les chiens domestiques
- 06-Comment ça marche, la sélection naturelle ?
- 07-La sélection naturelle et le lignage humain.
- 08-Les bases de la variation héréditaire, première partie
- 09-Les bases de la variation héréditaire, deuxième partie.
- 10-De la variation à la spéciation (1)
- 11-De la variation à la spéciation (2)
- 12-De la variation à la spéciation 3
- 13-De la variation à la spéciation (4)
- 14-Les génomes sont comme des anciens textes (1)
- 15-Les génomes comparés aux textes anciens (2)
- 16-Les génomes comparés aux textes anciens (3): les origines de l'homme
- 17-Le génome comparé à un texte ancien (4)
- 18-Le génome comparé à un texte ancien (5): rattacher le tout ensemble. (Cet article)
- 19-Les arbres généalogiques des espèces, des gènes, et tri incomplet des lignées
- 20-Tri de lignage incomplet et taille d’une population ancestrale
- 21-Une introduction à l’homoplasie et à la convergence évolutive
- 22-Evolution convergente et homologie profonde.
- 23-Coévolution et la course à l’armement prédateur / proie
- 24-Le parasitisme, le mutualisme et la co-spéciation
- 25-Comprendre l’endosymbiose
- 26-La diversification cambrienne et la mise en place des plans d’organisation animaux. Première partie.
- 27-La diversification cambrienne et la mise en place des plans d’organisation animaux. 2e partie.
- 28-La mise en place des plans d’organisation des corps vertébrés, première partie.
- 29-La mise en place des plans d’organisation des corps vertébrés, deuxième partie.
- 30-La mise en place des plans d’organisation des vertébrés : Troisième partie.
- 31-La mise en place des plans d’organisation des vertébrés, quatrième partie.
- 32-Des reptiles aux mammifères.
- La révolution placentaire : deuxième partie.
- Du primate à l’humain, première partie.
- Du primate à l’humain, deuxième partie
- Du primate à l'humain (3)
- La paléontologie hominienne : une petite esquisse des preuves actuelles
- Devenir humain (1) : Eve mitochondriale et Adam Y-chromosomique
- Analogie entre évolution biologique et évolution du langage
- Devenir humain (3) : paléogénomiques et la toile emmêlée de la spéciation humaine
- 41-L’évolution, Partie 1: frontières scientifiques, abiogenèse et apologétique chrétienne
- 42-Aux frontières de l’évolution, Partie 2: l’abiogenèse et la question du naturalisme
- 43-Aux frontières de l’évolution, Partie 3 : l’hypothèse du monde à ARN
- 44-Aux frontières de l’évolution, partie 4 : Contingence versus convergence
- 45-Aux frontières de l’évolution, partie 5 : Contigence versus convergence dans l’expérience LTEE
- 46- L’évolution et le chrétien, première partie: Est-ce que l’évolution est un mécanisme sans but ?
- 47- L’évolution et le chrétien, deuxième partie: Une créature merveilleuse
- 48- L’évolution et le chrétien, partie 3 : Dire la vérité dans l’amour




